
Reports of research work funded by grants prior to 2016
Institute of Environmental Science & Research Ltd
Dissecting the New Zealand meningococcal B epidemic using whole genome sequencing
X Ren
Invasive Pathogens Laboratory
In this study we set out to use whole genome sequencing to understand the emergence and adaptation of the New Zealand serogroup B epidemic meningococci strain (NZMenB). The aim of the study was to expand on our previous studies to sequence more isolates from key periods during the epidemic to understand emergence and possible vaccine adaptation. This project started in late October 2015, so far, we spent most of our time selecting, extracting DNA and sequencing meningococci isolates. We have finished first batch of whole genome sequencing and are in the process of analysing the data generated to see if more targeted sequencing are needed.
Aim 1: Use WGS to understand NZMenB emergence
We proposed to sequence and analyse an additional 30 NZMenB isolates from 1991 to 1993 as well as 18 pre-1991 serogroup B isolates. We also proposed to use MLST and porA PCR to analyse isolates from 1980s (excluding those identified as the Serogroup A epidemic type) to identify those from the ST-41/44 clonal complex and investigate whether the epidemic porA type existed before the epidemic.
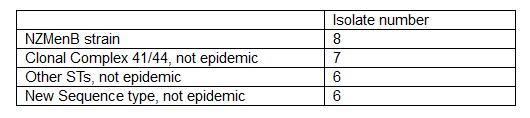
So far we have concentrated in analysing and sequencing pre 1991 isolates. A summer research student organised and attempted to identify porA allele from 100 meningococci isolates from 1987 to 1991. Of these reactions only 42 was successful and epidemic porA was not found. For whole genome sequencing, we also tried to grow 82 isolates from 1988 to 1991 for genomic DNA extraction however only 16 grew. Apparently a freezer failure in 1991 may have affected the viability of these isolates. We also organised previously extracted DNA samples and selected 11 more for WGS based on previous typing information. We extracted fine typing and MLST from these 27 isolates (Table 1).
Detailed analysis of the sequences is still ongoing. So far increasing the number of 1991 isolates has strengthened our initial finding that the diversity of the NZMenB was already present early in the epidemic suggesting the strain has been circulating long before the epidemic started. We will report the detailed analysis in the final report of this project.
Aim 2: Adaption of the NZMenB strain to vaccination
We proposed to sequence 48 more isolates from 2002, 2006, 2007 and 2010. We sequenced 13 isolates from 2002, 12 from 2006, 13 from 2007 and 10 from 2010. Among these isolates, there were 31 ST-42, 12 ST-154, 2 new STs and 4 other STs. Adding these isolates to previous analysis resulted in 32 pre-vaccination and 39 post-vaccination ST-42 isolates. Recently a prokaryote specific GWAS tool named bug was released (https://github.com/sgearle/bugwas). We tried this programme on our data set. We analysed the data in two ways, based on the core single nucleotide polymorphisms and indels (insertions and deletions) and based on the whole genome using a kmer approach. The programme did find principal component that can separate pre-vaccination vs post-vaccination isolates and about 10 SNPs that were above the noise but failed multiple test correction, which suggest the number of samples are still insufficient. With other less stringent corrections, several SNPs did pass significance. These SNPs were the same ones that were found before using the smaller dataset. Currently we are working with epidemiologists at ESR to identify isolates from individuals that have been vaccinated. These isolates will be used in future studies. Detailed analysis is still ongoing, we will present this result in the final report of the project.


