
Reports of research work funded by grants prior to 2012
Malaghan Institute of Medical Research
Impact of Urate-lowering Therapy on Monocyte/macrophage phenotype in Gouty Arthritis
JL Harper
Background
The key risk factor for gout is elevated serum uric acid (hyperuricaemia). Although uric acid has been identified as a strong modifier of the adaptive immune response, limited work has been undertaken to investigate the specific effect of hyperuricaemia on the innate inflammatory cells associated with inflammation in gout.
Immune cells of the monocyte/macrophage cell lineage play a pivotal role in gouty inflammation. These cells are involved in all stages of disease progression, from triggering MSU crystal-induced inflammation in an acute attack via the production of proinflammatory cytokines such as IL1b and IL-6 to switching to produce TGFb during resolution. Based on our earlier clinical data monocytes from gout patients with hyperuricaemia produce significantly less proinflammatory cytokines compared to monocytes from healthy volunteers. These differences indicated that hyperuricaemia may directly alter monocyte function.
Aim
To determine how uric acid directly influences the MSU-induced proinflammatory response of human blood monocytes.
To compare the inflammatory response of monocytes from gout patients before and after Uric Acid-lowering therapy (ULT).
Results
In this study we isolated blood monocytes from normouricaemic healthy volunteers and measured the levels of proinflammatory cytokines produced induced by the stimulus lipopolysaccharide, in the presence of high levels of uric acid. As shown in Figure 1 LPS-stimulated monocytes treated with uric acid ex vivo produced lower levels of proinflammatory cytokines.
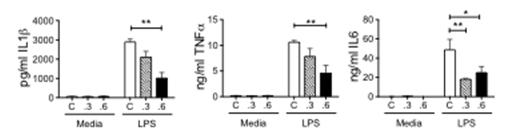
Figure 1. LPS-stimulated blood monocytes from healthy volunteers produce lower levels of pro-inflammatory cytokines in the presence of 0.3 and 0.6 mM uric acid.
Analysis of cellular proteins by western blotting showed rapid dephosphorylation of the MAPkinase signalling molecules involved in raising the LPS proinflammatory response consistent with shutdown of the inflammatory signalling cascade. In addition to a decrease in proinflammatory cytokine production, UA was also shown to upregulate anti-inflammatory IL-10 production. Mice given the uricase inhibitor oxonic acid (i.p.) in combination with soluble uric acid (i.v.) also exhibited and increase in IL-10 and a decrease in proinflammatory cytokine levels in the serum compared to control mice following LPS challenge (i.v.).
To look at the effect of lowering clinical hyperuricaemia, we also tested proinflammatory cytokine production by monocytes purified from a hyperuricaemic patient before and after ULT using the xanthine oxidase inhibitor allopurinol. ULT resulted in increased production of proinflammatory cytokines (e.g. TNFa and IL-12).
Conclusion
The findings of this study show that soluble UA may act to moderate inflammation induced by micropbial products such as LPS by shutting down proinflammatory signalling pathways and switching monocyte cytokine production towards anti-inflammatory IL-10. This would serve to regulate systemic inflammation and avoid inappropriate cytokine ‘storms’ associated with life threatening inflammation associated with sepsis.


