Reports of research work funded by grants prior to 2016
Victoria University of Wellington
Investigating the Anti-pain and Anti-inflammatory Effects of Novel Endocannabinoid-like Lipids
BM Kivel
School of Biological Sciences, Centre for Biodiscovery
Chronic pain, post-operative pain and pain resulting from cancer and arthritis negatively impact on the quality of life for pain sufferers and are a major economic burden to New Zealand. Particularly with New Zealand’s ageing population. Approximately 16% of New Zealanders currently suffer from chronic pain which is poorly treated with current medications. Drugs activating the mu-opioid receptor such as morphine and fentanyl, while effective at treating acute pain, become less effective over time and require higher doses to be administered to be effective (tolerance), cause constipation, respiratory depression and are addictive. Chronic use can also potentiate pain. Therefore, more effective anti-pain pharmacotherapies with reduced side effects and lower abuse potential would be of enormous benefit to millions of people worldwide.
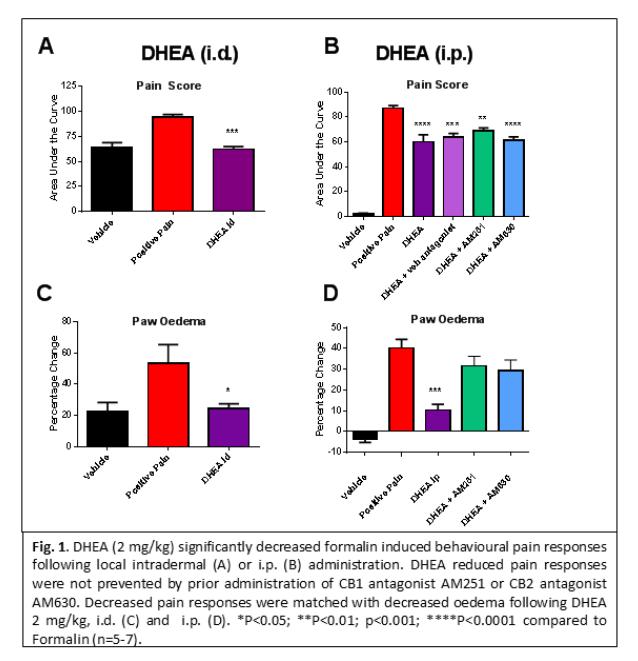
Bioactive lipids, similar to endogenous cannabinoids are known to modulate pain and inflammation. Therefore, our study chose to preclinically evaluate new bioactive lipids for their ability to modulate pain and inflammation. We utilised a mouse model of acute inflammatory pain, whereby, 2% formalin was injected into the hind paw of the mouse and the severity of pain behaviours and inflammation evaluated by an observer blinded to the treatment groups. We show that the bioactive lipid docosahexaenoyl ethanolamide (DHEA), significantly reduced both nociceptive pain (phase 1, between 0-10 min) and inflammatory pain behaviours (phase 2, 15-60 min). This effect was significant following both local intradermal (i.d.) footpad administration of DHEA (2 mg/kg) (Fig. 1A) and intraperitoneal (i.p.) administration (2 mg/kg) (Fig 1B). Data shows that DHEA was systemically active and not just effective following local administration, an improvement over many bioactive lipids which are rapidly inactivated and only act locally. Additional evaluation of footpad tissue taken at 60 min following 2% formalin and DHEA administration (2 mg/kg, i.p.) revealed a significant reduction in neutrophil numbers compared to formalin/vehicle treated controls. Significantly reduced footpad swelling (oedema) further confirmed DHEA’s anti-inflammatory effects (Fig. 1 C &D). All data passed the Shapiro-Wilk test for normality, therefore One Way ANOVA followed by Bonferroni’s multiple comparisons test were used to test multiple groups (area under the curve analysis of formalin pain behaviour and oedema). Two-way repeated measures ANOVA was used for time course data of formalin pain, motor coordination, temperature and tail withdrawal tests followed by Bonferroni’s multiple comparisons tests. Data was considered statistically significant when (p<0.05).
Because DHEA is a bioactive lipid, it was expected to act via cannabinoid receptor activation. To investigate the mechanism underlying both the analgesic and anti-inflammatory effects of DHEA we pre-treated a group of mice with either the cannabinoid 1 receptor (CB1) antagonist AM251 or CB2 antagonist AM630 followed by DHEA (2 mg/kg/i.p.) and evaluated behavioural responses in the formalin-induced inflammation model. There was no significant change in the behavioural pain scores in either phase 1, nociceptive pain, or phase 2, inflammatory pain, with either cannabinoid antagonist. Measures of oedema (footpad swelling) also showed no significant reduction in inflammation following either CB1 or CB2 antagonist administration compared to formalin/vehicle treated controls. Combined, this data suggests that the analgesic and anti-inflammatory effects of DHEA are not mediated via activation of cannabinoid receptors. The exact mechanism of action remains to be determined.
The warm-water tail withdrawal assay activates a thermal nociceptive response mediated by neurones in the central nervous system (CNS). DHEA (2 mg/kg) showed no change in withdrawal latency compared to vehicle treated controls suggesting that DHEA is not centrally active. Therefore, additional evaluation of CNS penetrance of DHEA was undertaken. Mice were administered C13 labelled DHEA (2 mg/kg, i.p) or vehicle and brain tissue was taken following sacrificed at either 10 or 30 min. Liquid chromatography mass spectroscopy analysis revealed C13 labelled DHEA was present in the brain at both 10 and 30 min (at low ng/ml levels). Full quantification experiments are ongoing (n=2 per group). This suggests the DHEA’s lack of effect in the CNS is not due to its inability to cross the blood-brain barrier, or to rapid inactivation but likely due to its specific modulation of inflammatory pain rather than nociceptive pain.
To evaluate the side effect profile of DHEA we also measured rectal temperature and used the rotarod test of motor coordination to evaluate whether DHEA had sedative effects, a common side-effect of many pain medications. DHEA had no effect on rectal temperature or the latency to fall in the accelerating rotarod test at the therapeutic dose
of (2 mg/kg) at any time point between 5-120 min.
We can conclude that DHEA at a dose of 2 mg/k is effective in attenuating acute inflammatory pain in the mouse. This anti-inflammatory effect is confirmed by reduced footpad oedema and reduced neutrophil infiltration into the footpad tissue. In addition, DHEA showed no sedative effects. The mechanism of how DHEA exerts its anti-inflammatory effects are unknown, but it is not due to modulation of either CB1 or CB2 receptors, as antagonist administration failed to attenuate the effects of DHEA. Bioactive lipids, such as DHEA hold promise for the development of therapeutics to treat inflammation and inflammatory pain, particularly as it is effective following both systemic and local administration and is devoid of sedative effects.