
Reports of research work funded by grants prior to 2012
Malaghan Institute of Medical Research
The role of M1 and M2 macrophages in MSU crystal-induced inflammation in gout
JL Harper
Background
Gouty arthritis is an inflammatory condition driven by the formation of monosodium urate crystals (MSU) in the joints and connective tissues. During the progression of the inflammatory response, infiltrating monocytes have been shown to differentiate into a hyper-inflammatory macrophage (M1-like) phenotype. In vivo stimulation of this M1 macrophage with MSU crystals enhances the inflammatory response indicating that this macrophage may exacerbate inflammation during a gout attack.
The colony stimulating factors M-CSF and GM-CSF exert significant influence over the differentiation pathway of the monocyte/macrophages. Monocyte exposure to M-CSF or GM-CSF is reported to result in the development of resident or inflammatory macrophage phenotypes respectively. Our earlier data showed that there is a temporal shift in CSF production in vivo where early recruited monocytes are exposed to high levels of M-CSF that is then followed by uptake of GM-CSF. This pattern of CSF production supports our reported observation that the MSU crystal recruited monocyte/macrophage shifts from a non-inflammatory to hyper-inflammatory phenotype in vivo.
Aim
To investigate the inflammatory phenotype of bone marrow-derived M-CSF and GM-CSF generated macrophages following stimulation with the gout-causing agent MSU in vitro.
This work was expanded to investigate the impact of the anti-inflammatory molecule TGFb1, a pivotal cytokine produced during the resolution of a gout attack, affected the M-CSF and GM-CSF macrophage inflammatory responses.
Results
Analysis of the cell surface markers on the two CSF macrophages identified GM-CSF macrophages (GMM) as expressing lower levels of the classical macrophage surface markers F4/80, CD14 and M-CSF receptor compared with M-CSF macrophages (MM). The GMM produced high backgound levels of IL-1b compared to MM. MSU stimulation triggered IL-1b production by MM but not GMM. However, upon exposure to TGFb1 GMM produced significantly greater levels of IL-1b whereas MM decreased IL-1b production (Figure 1).
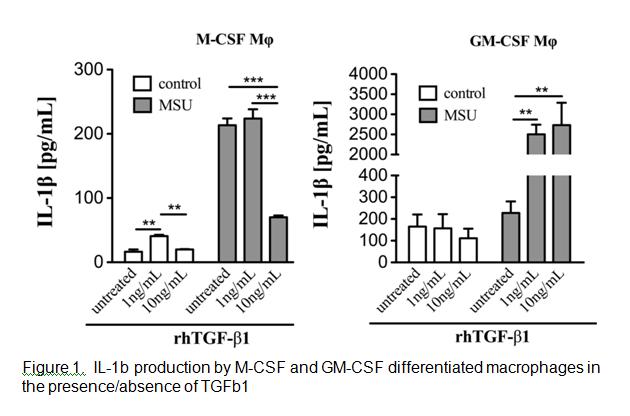
Conclusion
These data indicate that M-CSF resident macrophage phenotype is primarily responsible for initiating the MSU induced inflammatory response in gout and is shut down by the generation of TGFb1. Over time GM-CSF driven differentiation of infiltrating monocytes results in the development of an M1 macrophage phenotype that is further augmented by the presence of TGFb1. These findings serve to illustrate the differential effects of TGFb1 on different macrophage phenotypes and the potential for TGFb1 to either drive or shutdown inflammation.


